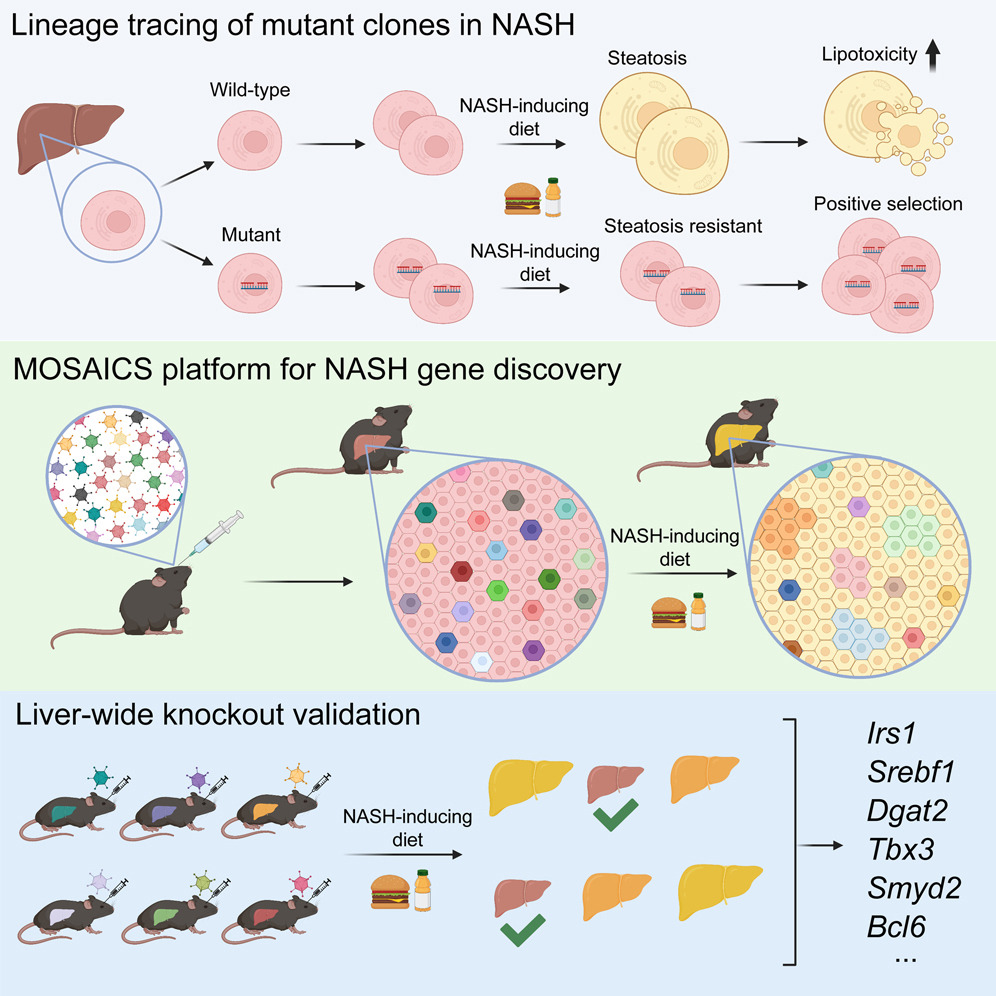
 Somatic mutations in nonmalignant tissues accumulate with age and injury, but whether these mutations are adaptive on the cellular or organismal levels is unclear. To interrogate genes in human metabolic disease, we performed lineage tracing in mice harboring somatic mosaicism subjected to nonalcoholic steatohepatitis (NASH). Proof-of-concept studies with mosaic loss of Mboat7, a membrane lipid acyltransferase, showed that increased steatosis accelerated clonal disappearance. Next, we induced pooled mosaicism in 63 known NASH genes, allowing us to trace mutant clones side by side. This in vivo tracing platform, which we coined MOSAICS, selected for mutations that ameliorate lipotoxicity, including mutant genes identified in human NASH. To prioritize new genes, additional screening of 472 candidates identified 23 somatic perturbations that promoted clonal expansion. In validation studies, liver-wide deletion of Tbx3, Bcl6, or Smyd2 resulted in protection against hepatic steatosis. Selection for clonal fitness in mouse and human livers identifies pathways that regulate metabolic disease.
Somatic mutations in nonmalignant tissues accumulate with age and injury, but whether these mutations are adaptive on the cellular or organismal levels is unclear. To interrogate genes in human metabolic disease, we performed lineage tracing in mice harboring somatic mosaicism subjected to nonalcoholic steatohepatitis (NASH). Proof-of-concept studies with mosaic loss of Mboat7, a membrane lipid acyltransferase, showed that increased steatosis accelerated clonal disappearance. Next, we induced pooled mosaicism in 63 known NASH genes, allowing us to trace mutant clones side by side. This in vivo tracing platform, which we coined MOSAICS, selected for mutations that ameliorate lipotoxicity, including mutant genes identified in human NASH. To prioritize new genes, additional screening of 472 candidates identified 23 somatic perturbations that promoted clonal expansion. In validation studies, liver-wide deletion of Tbx3, Bcl6, or Smyd2 resulted in protection against hepatic steatosis. Selection for clonal fitness in mouse and human livers identifies pathways that regulate metabolic disease.
Read the full article in Cell.