
Full Bio

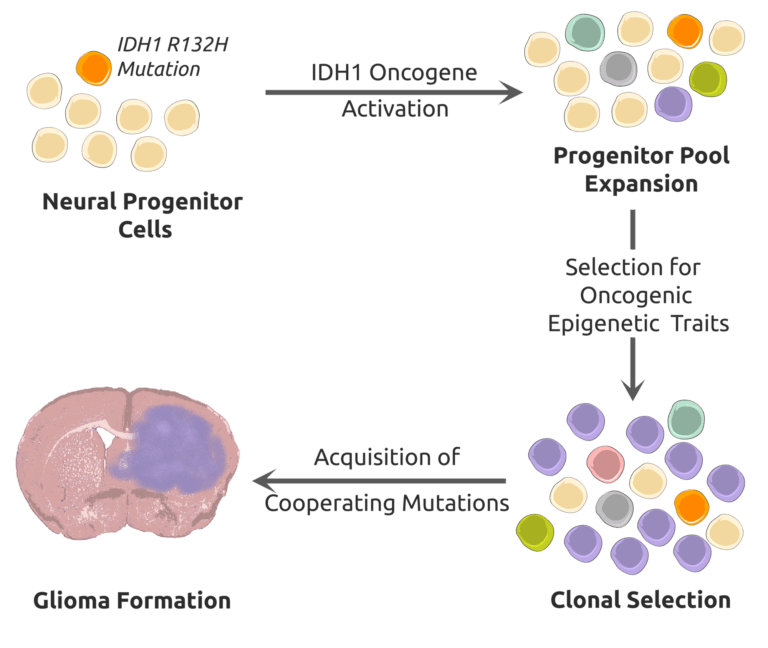
IDH1 mutations are the signature genetic feature of lower grade gliomas and secondary glioblastomas. They are thought to initiate gliomagenesis by causing accumulation of the oncometabolite (R)-2-hydroxyglutarate in neural progenitor cells. (R)-2-hydroxyglutarate, in turn, controls the activity of dioxygenase enzymes which regulate chromatin structure, hypoxia signaling, and other key aspects of neural cell biology. Collectively, these effects promote brain tumor initiation. Although this framework represents a significant advance in our understanding of the oncogenicity of IDH1 mutations, detailed characterization of the molecular cascades that link (R)-2-hydroxyglutarate accumulation with gliomagenesis has not been fully completed.
We are currently undertaking complementary top-down and bottom-up approaches to dissect specific oncogenic mechanisms engaged by IDH1 mutations. To systematically identify critical proximal effectors of (R)-2-hydroxyglutarate in glioma, we are performing CRISPR/Cas9 screens to uncover dioxygenases that control malignant transformation in cellular contexts that closely recapitulate glioma genetics. We are also using a novel genetically engineered mouse model of glioma created in our laboratory to characterize the dynamics of the distal effects of mutant IDH1 on the epigenome and the transcriptome. Taken together, findings from these studies are expected to provide a comprehensive understanding of how (R)-2-hydroxyglutarate induces glial cell transformation in vivo. These findings may reveal metabolic mechanisms of transformation with relevance beyond the setting of IDH1 mutant glioma. Furthermore, our findings may reveal unappreciated therapeutic opportunities to impede brain tumor progression.
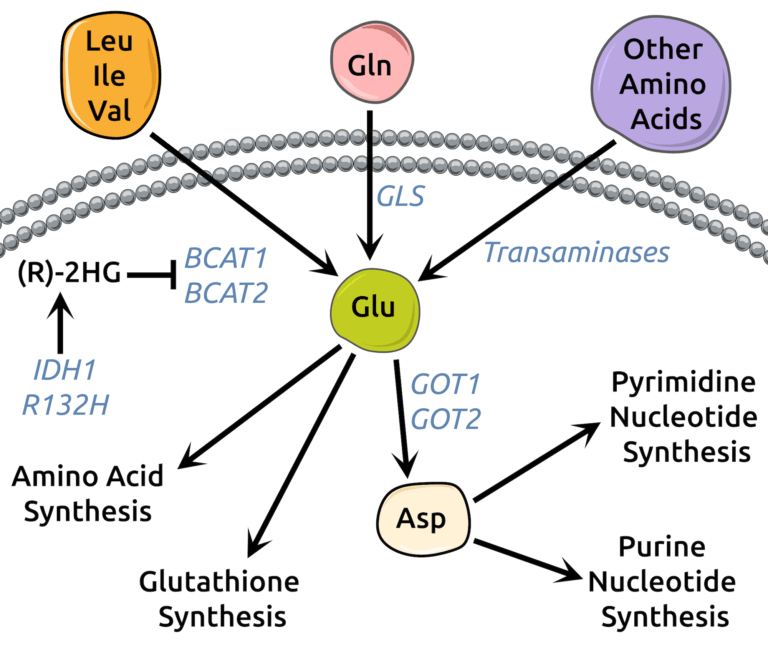
The paradigm of malignant transformation by IDH1 mutations holds that (R)-2-hydroxyglutarate produced by IDH1 mutant enzymes directly modulates the activity of oncogenic or tumor suppressive dioxygenase enzymes to promote tumorigenesis. Recently, we showed that (R)-2-hydroxyglutarate can also regulate the activity of another class of enzymes known as transaminases (Cell 175, 101-116, 2018). Specifically, we found that (R)-2-hydroxyglutarate directly inhibits the branched chain amino acid transaminases BCAT1 and BCAT2. These enzymes play central roles in nitrogen metabolism in glial cells, and our work revealed that (R)-2-hydroxyglutarate accumulation impairs the BCAT-dependent synthesis of nitrogenous metabolites.
These findings provide a mechanistic explanation for metabolic differences observed between IDH1 mutant and wild-type brain tumors but, at the same time, prompt fundamental questions about nitrogen metabolism programs in cancer. How do tumor cells couple the catabolism of specific amino acids to the synthesis of key nitrogenous metabolites? How do tumor cells engage compensatory amino acid catabolism pathways to adapt to nitrogen limitation? The answers to these questions have been obscured by conventional depictions of metabolic pathways from carbon-centric standpoints. We aim to answer these questions using metabolomic profiling and isotope tracing approaches in in vitro and in vivo glioma models to systematically map nitrogen metabolism pathways. These studies are expected to illuminate novel patterns of nitrogen incorporation in IDH1 mutant brain tumors as well as other cancers that display BCAT-independent metabolic phenotypes.
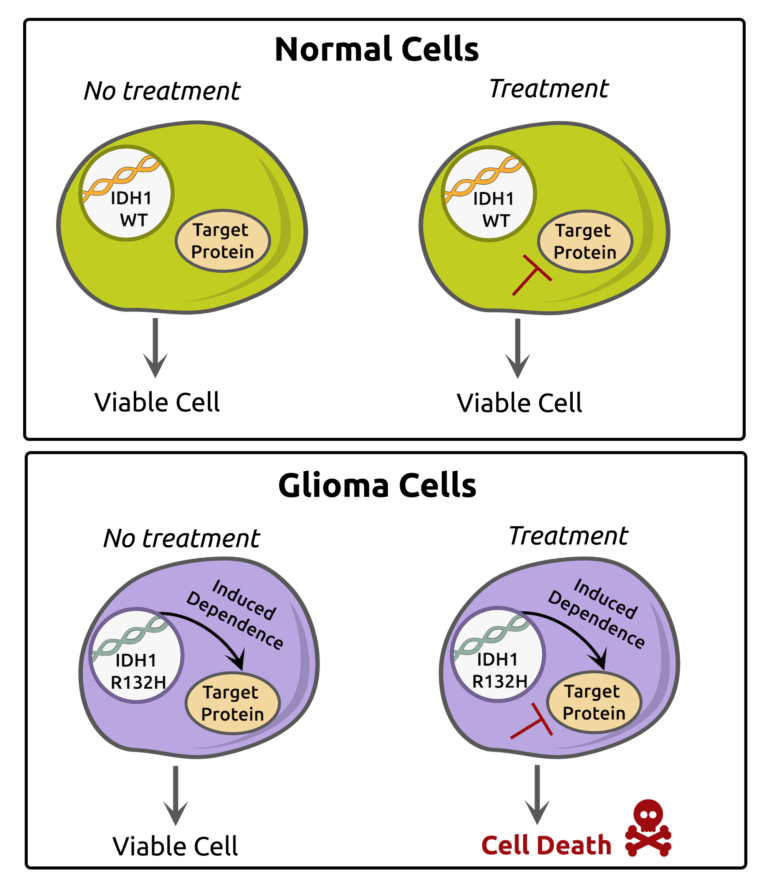
Malignant gliomas are notoriously refractory to therapy, and there is a dire unmet need for new treatments. Discovery of the high prevalence of IDH1 mutations in lower grade gliomas and secondary glioblastomas has opened new avenues for therapeutic intervention, including the use of direct inhibitors of mutant IDH1 enzymes. An alternative approach to treating these brain tumors entails the exploitation, rather than inhibition, of IDH1 mutant enzymes through the discovery of associated synthetic lethal interactions. Our previous work describing nitrogen metabolism reprogramming by IDH1 mutations led to the development of a new synthetic lethality-based treatment strategy that is currently being tested in a Phase I trial for glioma patients (NCT03528642).
We are pursuing both hypothesis-driven and unbiased approaches to identify additional synthetic lethal interactions with the canonical IDH1 R132H oncogene. Because radiation is a cornerstone of the standard-of-care treatment protocol for glioma, we are particularly interested in discovering collateral vulnerabilities induced by IDH1 mutations that impact radiosensitivity. To evaluate the translational relevance of our findings, we use patient-derived and genetically engineered mouse models of glioma to conduct preclinical efficacy studies. Our long-term goal is to lay the basic and translational scientific groundwork needed to support clinical testing of new glioma treatment strategies using IDH1 mutations as predictive biomarkers.



















